3.1. Виробництво стерильної продукції
Виробництво стерильної продукції залежно від способу досягнення стерильності GMP ВООЗ ділять на наступні категорії:
- виробництво, при якому продукція кінцево стерилізується в герметизованій первинній упаковці;
- виробництво, при якому препарати стерилізуються фільтрацією;
- виробництво в асептичних умовах із стерильних вихідних речовин та матеріалів.
GMP ЄС технологічні процеси підрозділяє на дві категорії:
- виробництво, при якому продукція кінцево стерилізується в укупореній (герметично) первинній упаковці;
- виробництво в асептичних умовах на деяких або на всіх стадіях.
Приклади технологічних операцій, що вимагають різних класів
|
|

|
|

Лікарські засоби для парентерального введення великого об’єму необхідно фасувати на робочому місці з ламінарним потоком повітря (клас А) при відповідній якості повітря навколишнього середовища класу С.
Загальні вимоги. Стерильні лікарські засоби (ЛЗ) необхідно виготовляти в чистих зонах. Доступ персоналу і/чи вихідної сировини, матеріалів, напівпродуктів обладнання в чисті приміщення дозволяється тільки через повітряні шлюзи. В чистих зонах необхідно підгримувати належну чистоту, що регламентується вимогами GMP, а повітря, що до них подається, повинно проходити очищення через фільтри відповідної ефективності.
Допоміжні операції (підготовка вихідної сировини і матеріалів, виготовлення напівпродуктів, фасування в первинну упаковку, стерилізація) необхідно проводити в окремих зонах чистого приміщення.
Необхідну ступінь чистоти повітря необхідно забезпечувати, базуючись на результатах валідації.
Згідно з вимогами GMP ВООЗ чисті приміщення для виробництва стерильної продукції класифікує у відповідності вимог до характеристик на класи чистоти А, В, С і Д.
Вимоги до технологічного процесу
На всіх стадіях технологічного процесу, включаючи стадії попередньої стерилізації, необхідно проводити заходи, що зводять до мінімуму ризик контамінації.
Система класифікації повітря при виробництві стерильної продукції
|
|
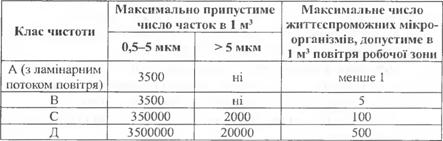
Для зон, що мають клас чистоти А, В і С, система товарозабезпечення повинна мати відповідні фільтри як, наприклад фільтри НЕРА.
Клас А: Локальні зони для технологічних операцій, що потребують самого мінімального ризику контамінації (зони наповнення, укупорки, відкриття ампул та флаконів, змішування в асептичних умовах).
Клас В: Навколишнє середовище для зони А у випадках приготування і наповнювання в асептичних умовах.
Клас С та Д: Чисті зони для проведення технологічних операцій, які припускають більший ризик контамінації, при виробництві стерильної продукції.
Для підтвердження класу чистоти зон в робочому стані необхідно періодично проводити мікробіологічний контроль.
1. Препарати, що містять живі мікроорганізми, забороняється виробляти та фасувати в приміщеннях, де будуть виготовляти інші лікарські засоби.
2. Валідація технологічних процесів, що проводяться в асептичних умовах, повинна включати модулювання процесу з використанням живильних середовищ через встановлені проміжки часу.
Рівень контамінації не повинен викликати ріст мікроорганізмів на живильних середовищах: припускається ріст менше, ніж 0,1% випадків з довірчою вірогідністю 95% (норма GMP ЄС).
3. Проведення валідації не повинне негативно впливати на хід технологічного процесу та якості продукції.
4. Джерело води, обладнання для обробки води та оброблена вода постійно контролюються на хімічну та мікробіологічну контамінацію. Наслідки контролю протоколюються.
5. Вихідна сировина та матеріали повинні мати мінімальну контамінацію. ,
6. Ємність та матеріали, що утворюють волокна, що віддаляються від них самочинно, виключаються з використання.
7. Інтервали часу між миттям, сушкою, стерилізацією первинної упаковки, ємностей та обладнання, а також інтервали часу між стерилізацією їх та використанням повинні бути мінімальними.
8. Інтервали часу між стадіями технологічного процесу повинні бути мінімальними.
9. Будь який газ, що контактує к ісхнолої ічному проікч і і рімчи нами або іншою продукцією, повинен пройти стерилізуючу фільтрацію (GMP ВООЗ).
10. Ефективність нових методик та способів ведення технологічного процесу необхідно стверджувати валідацією, яку проводять регулярно.
Віимоги до стерилізації
Загальні вимоги.
1. Стерилізація може проводитись з використанням вологого (пари) або сухого шару (нагрітого повітря), етилен оксиду або іншої відповідної газоподібної речовини, стерилізуючою фільтрацію з наступним фасуванням в асептичних умовах, а також з використанням радіоактивного випромінювання. Кожний метод має свої позитивні сторони, область використання та гранічні вимоги. Як правило, перевагу віддають термічним способам стерилізації (при умові, що вони можуть бути прийнятими для даної продукції (СіМР ВООЗ)).
2. Всі способи стерилізації повинні пройти валідацію. Особливу увагу необхідно приділяти методам, які не є фармакопейними або не відповідають способам, наведеним у галузевих нормативних документах. У всіх випадках використаний метод стерилізації повинен відповідати реєстраційним та ліцензійним документам.
3. Перед вибором методу стерилізації необхідно ствердити фізичними та біологічними методами придатність його використання для даної продукції та що при його використанні створюються належні умови стерилізації у всіх місцях (частинах) продукції, що стерилізується.
4 Для ефективної стерилізації необхідно піддавати відповідній обробці всю вихідну сировину, матеріали та напівпродукти.
5. Біологічні індикатори необхідно розглядати як допоміжний метод контролю за процесом стерилізації. У випадку використання біологічних індикаторів необхідно вживати суворі заходи обережності, попереджуючи мікробну контамінацію із самих індикаторів.
6. Нестерильну продукцію і продукцію, що подається на стерилізацію, необхідно чітко диференціювати та ідентифікувати за допомогою відповідного маркування.
7. кожний цикл термічної стерилізації фіксувати (на діаграмі часу) з використанням обладнання, що має достатню точність та чіткість, як допоміжні індикатори за контролем температури, можуть бути використані хімічні або біологічні.
8. При проведенні валідацй' враховувати період, за який вся маса завантажена в стерилізатор, і температуру, що забезпечує гарантування стерильності.
9. При стерилізації вологим жаром необхідно контролювати температуру і тиск.
10. Необхідно контролювати, щоб для стерилізації використовувалась пара належної якості, що не містить домішок, які могли б стати причиною контамінації продукції або обладнання.
11. Технологічний процес стерилізації сухим жаром має передбачати циркуляцію повітря, що подається в камеру і підлягає стерилізуючій фільтрації пропусканням через фільтри НЕРА.
12. Радіаційна стерилізація використовується головним чином для стерилізації матеріалів і продукції, що чутлива до нагрівання.
13. Для стерилізації можуть бути використані гази і фуміганти. Але необхідно довести, що залишкова концентрація їх знаходиться в нормі відповідно до специфікації на стерильну продукцію та матеріали
Примітка ОМ Р наголошує, що при використанні стерилізуючої фільтрації необхідно враховувати:
чи підтримує лікарський засіб ріст будь-яких мікроорганізмів; чи змінює лікарський засіб характеристики фільтруючого матеріалу;
^ чи є достатнім досягнення стерильності продукції з використанням фільтра з пропускною здатністю 0,22 мкм для видалення мікроорганізмів.
Принципи та правила виробництва лікарських засобів, у тому числі стерильних для людини введені в Європейському Союзі законодавчо Директивою Комісії ЄС 91/356/ЄЄС “Про встановлення основних принципів і правил належної виробничої практики лікарських засобів для людини” і є обов’язковими для всіх країн СС, а також для третіх країн, що експортують лікарські засоби.
Належна виробнича практика необхідна не тільки для забезпечення якості лікарських засобів, але й як засіб для усунення технічних бар’єрів торгівлі між країнами ЄС, а також третіми країнами.
Тому виконання всіх принципів і правил, що включені до порадника GMP (GoodManufacturer Practice), включені до порадника СС і е обов’язковими.
В Директиві 91/356 ЄЄС приведені визначення GMP, як “частини забезпечення якості, яка гарантує, що продукцію постійно виробляють і контролюють за стандартом якості, що відповідає її призначенню”, “Забезпечення якості лікарських засобів (pharmaceutical quality assurance) - сукупність всіх організаційних заходів, прийнятих з метою забезпечення якості лікарських засобів, що визначаються їх призначенням”. Тобто забезпечення якості включає не тільки належну виробничу практику, а й інші види діяльності, які виходять за межі запропонованого в ЄС посібника з GMP, але тільки пов’язані з GMP.
Наприклад, виробник повинен регулярно удосконалювати способи виробництва відповідно з досягненнями науково-технічного процесу. Якщо необхідно внести зміни до ліцензійного досьє, то заявка на зміни повинна бути направлена в компетентні уповноважені органи.
Вимоги до виробництва стерильних лікарських засобів. Принципи
При виробництві стерильної продукції необхідно дотримуватись спеціальних правил, що висувають вимоги до виробничого процесу з метою зведення до мінімуму ризику контамінації мікроорганізмами, механічними частками та пірогенними речовинами. Дотримання цих правил залежить в першу чергу від належного фаху, рівня знань і виробничої дисципліни персоналу Виробництво і контроль якості необхідно проводити в чіткій відповідності з розробленими та валідовани- ми методами виробництва і контролю.
Жодна операція або випробування, включаючи технічний процес чи контроль якості ютового продукту, не можуть розглядатись в якості єдиного фактора, що гарантує та засвідчує стерильність або показники якості готового продукту.
Наведені нижче правила не змінюють, а доповнюють правила належної виробничої практики і є специфічними вимогами до виробництва та контролю якості стерильної продукції.
Вимоги до укупорки стерильної продукції
1 Первинні упаковки з разфасованою продукцією повинні бути укупорсні і герметизовані належним засобом, що пройшов валідацію.
Скляні або пластикові ампули у 100% випадків необхідно перевіряти на цілісність і герметичність. Від інших видів продукції необхідно відбирати проби для контрольних випробувань первинних упаковок відповідно до стандартних робочих методик.
2. Первинні упаковки, укупорені під вакуумом, через відповідні (установлені) проміжки часу необхідно перевіряти на збереження вакууму.
3. Первинні упаковки з лікарськими засобами для парентерального введення необхідно контролювати поштучно на сторонні включення або інші дефекти. Всі методи контролю механічних включень повинні пройти валідацію перевірки, її наслідки протоколюються.
Вимоги до контролю якості
1. Визначення стерильності готової продукції необхідно розглядати тільки як завершений етап в комплексі контрольних досліджень, що проводяться з метою гарантування і достовірності стерильності. Дослідження повинні пройти валідацію для кожної конкретної продукції.
2. У випадках, коли було санкціоновано зниження вимог до параметрів, особливу увагу необхідно приділяти валідації і контролю всього виробничого процесу в цілому![]()
3. Зразки готової продукції, відібрані для контролю на стерильність, повинні бути репрезентативними для всієї ссрії. Відбір проб необхідно проводити додатково із тих частин серії, для яких передбачається підвищений ризик мікробної контамінації:
для продуктів, виготовлених в асептичних умовах, відбір проб повинен включати первинні упаковки з ліками, виготовлені на початку та в кінці виготовлення серії, а також після передбаченої або непередбаченої тривалої перерви в роботі;
'Ґ для продукції, простерилізованої в герметичній первинній упаковці з використанням термічних засобів, відбір проб необхідно проводити із тих частин серії, які стерилізувались у найбільш холодних частинах камери.
4. Воду, напівпродукти та готову продукцію необхідно контролювати на ендотоксини із застосуванням фармакопейних методик. У випадку інфузійних розчинів великого об'єму такий контроль є обов’язковим і передбачається в технологічному регламенті. Його проводять завжди, додатково до контрольних визначень за вимогами і ліцензійної документації на готову продукцію.
1. Серії, що були відбраковані за наслідками перевірки на стерильність, не підлягають реалізації.
До стерильних ліків для ін’єкцій відносяться різні дисперсні системи: водні та олійні розчини, тонкі суспензії, емульсії, а також стерильні порошки і таблетки, які розчиняють перед введенням у стерильному розчиннику. Залежно від місця введення ліки для ін’єкцій поділяють на внутрішньошкірні. підшкірні, внутрішньом’язові, внутрішньосудинні, спинномозкові, внутрічерепні, внутрішньочеревні, внутрішньоплев- ральні, внутрішньосуглобні, що свідчить про широке розповсюдження цього способу введення.
Останнім часом спостерігається тенденція до зростання виробництва стерильних і асептично приготованих ліків. Разом з традиційними ліками для ін’єкцій і інфузій, а також очними краплями, фармацевтичні підприємства і установи готують великий асортимент офтальмологічних розчинів для зрошувань, використовуваних при мікрохірургії ока, дитячі рідкі ліки для внутрішнього і зовнішнього застосування. Це обумовлює необхідність особливих вимог до організації технологічного процесу виробництва стерильних ліків.
Значно розширилися наукові дослідження щодо розробки і вдосконалення технології і контролю якості стерильних ліків. Затверджені ряд методичних документів і НТД, що регламентують вимоги до цієї групи ліків. ВООЗ розроблені міжнародні вимоги (GMP), що висуваються до виробництва стерильних ліків (дотримуються більше 80 країнами світу). У фармацевтичну практику впроваджені нові технології, методи контролю і устаткування, які забезпечують високу якість стерильної продукції.
Нині існує велика номенклатура стерильних лікарських форм, випуск яких обчислюється в мільярдах упаковок. Питома вага лікарських препаратів для парентерального застосування складає близько 30—40 % від усіх готових лікарських засобів (ГЛЗ).
Згідно зі статтею Державної фармакопеї України “Лікарські засоби для парентерального застосування”, їх класифікують таким чином:
- ін’єкційні лікарські засоби;
- внутрішньовенні інфузійні лікарські засоби;
- концентрати для ін’єкційних або внутрішньовенних інфузійних лікарських засобів;
- порошки для ін’єкційних або внутрішньовенних інфузійних лікарських засобів;
- імплантанти.
Вимоги цієї статті не поширюються на препарати, виготовлені з людської крові, імунологічні і радіофармацевтичні препарати, протези для імплантації.
Ін’єкційні лікарські засоби - це стерильні розчини, емульсії або суспензії. Розчини для ін’єкцій повинні бути прозорими і практично вільними від частинок. Емульсії для ін’єкцій не повинні виявляти ознак розшарування. У суспензіях для ін’єкцій може спостерігатися осад, який повинен швидко диспергуватися при збовтуванні, утворюючи суспензію. Утворена суспензія повинна бути достатньо стабільною для того, щоб забезпечити необхідну дозу введення.
Внутрішньовенні інфузійні лікарські засоби — це стерильні водні розчини або емульсії з водою. Як дисперсійне середовище: вони повинні бути вільні від пірогенів та ізотонічні крові. Призначаються для застосування у великих дозах, тому не повинні містити антимікробних консервантів.
Концентрати для ін’єкційних або внутрішньовенних інфузійних лікарських розчинів являють собою стерильні розчини, призначені для ін’єкцій або інфузій після розведення. Концентрати розводять до вказаного об’єму відповідною рідиною перед застосуванням. Після розведення отриманий розчин повинен відповідати вимогам, що висуваються до ін’єкційних або інфузійних лікарських засобів.
Порошки для ін’єкційних або внутрішньовенних інфузійних лікарських засобів - це тверді стерильні речовини, поміщені в контейнер. При струшуванні з вказаним об’ємом відповідної стерильної рідини вони швидко утворюють або прозорий, вільний від частинок розчин, або однорідну суспензію. Після розчинення або суспендування вони повинні відповідати вимогам, що висуваються до ін’єкційних або інфузійних лікарських засобів.
Імплантати — це стерильні тверді лікарські засоби, що маюіь від- повідні для парентеральної імплантації розміри і форму, і вивільняючі речовини, що діють протягом тривалого часу. Вони повинні бути упаковані в індивідуальні стерильні контейнери.
Широкому застосуванню і виробництву препаратів для парентерального введення сприяє ряд переваг перед іншими ГЛЗ:
- швидка дія і біологічна доступність лікарської речовини;
- точність дозування;
- можливість введення лікарського препарату хворому, який знаходиться без свідомості;
- можливість заміни в організмі крові після значних її втрат;
- відсутність впливу секретів шлунково-кишкового тракту і ферментів печінки;
- стабільність і можливість зберігання протягом тривалого терміну;
- можливість створення запасів стерильної продукції, що полегшує і прискорює її відпуск з аптек, та ін.
Препарати для ін’єкцій заводського виробництва випускаються в контейнерах зі скла - ампулах, флаконах; прозорих пластмасових упаковках з полімерних матеріалів та шприц-тюбиках разового застосування.
Парентеральне застосування препаратів передбачає порушення шкірного покриву, що пов'язане з можливим інфікуванням і введенням механічних включень. Тому виробництво ін’єкційних лікарських форм порівняно з іншими (таблетки, мазі, супозиторії) має специфічні особливості, які диктуються вимогами до ін’єкційних розчинів (стерильність, апірогенність, відсутність механічних включень, стабільність і т.д., а Для деяких препаратів - ізотоніч- ність, осмоляльність (осмолярність), ізоіонічність, ізогідричність, певна іонна сила і в’язкість).
Виробництво парентеральних лікарських засобів повинне відповідати вимогам GMP (Належна виробнича практика), що викладені у Керівництві за якістю ВООЗ і Європейського Союзу, а також у Керівництві 42-01-2001,42-01-2003 і нормативних актах „Про організацію організації роботи аптечних та хіміко-фармацевтичних підприємств, 2003 р.”.
Визначення якості ампульного скла. Підготовка ампул до наповнення
Стерильна продукція випускається у спеціальних первинних упаковках, виготовлених із скломаси (ампули, флакони) або полімерних матеріалів (флакони, гнучкі контейнери, шприц-тюбики).
Упаковки для ін’єкційних препаратів поділяються на 2 групи:
- однодозові - такі, що містять певну кількість препарату, призначену для одноразової ін’єкції;
- багатодозові - такі, що забезпечують можливість багаторазового відбору з змності певної кількості препарату без порушення його стерильності.
Найбільш поширеним представником одноразової упаковки є ампула. Ампули - це скляні ємкості різної місткості і форми, що складаються з розширеної частини - корпуса і капіляра (стебла). У фармацевтичній промисловості найбільш поширені ампули місткістю 1,2,3,5 і 10 мл; 20 і 50 мл - характерні для ветеринарії. Капіляри ампул можуть бути рівними або з перетиском. Найбільш раціональними є ампули з псретиском, оскільки рідина з ампули не може потрапити в капіляр, що важливо при запаюванні і розтині ампул.
У нашій країні випускаються ампули для шприцевого і вакуумного наповнення.
До одноразових посудин відносять шприц-тюбик. Це тюбики з полімерних матеріалів з ін’єкційною голкою, захищеною ковпачком. Вони мають, як правило, спеціальне призначення і в різних країнах різну назву - цитола, майола, ампіна та ін.
Прикладом багатодозових посудин є флакони для інфузійних розчинів місткістю 50, 100, 250,500 мл, виготовлені зі скла або полімерних матеріалів. Перспективними посудинами для інфузійних розчинів є гнучкі контейнери, виготовлені з полівінілхлориду (ПВХ).
Скляні ємкості для ін’єкційних розчинів виготовляють з медичного скла, яке являє собою розчин (сплав) силікатів оксидів металів і деяких солей. Змінюючи склад компонентів та їх співвідношення, можна отримати скло із заданими властивостями.
Залежно від якісного і кількісного вмісту добавок, а також від набутих властивостей розрізняють 2 класи і декілька марок скла, що використовується у виробництві ампул.
Із 1996 року в Україні виробляються ампули зі скла медичного марки УСП-1 (ТУ У 480945-002), що за водостійкістю відповідає класу 1/121. В ампулах не допускається внутрішня залишкова напруга, яка створює питому різницю ходу променів більше 8-1 млн, чужорідні включення, сколи, забруднення, що не відмиваються, і скляний пил. Ампули УСП-1 повинні бути термічно стійкими і витримувати перепад температур не менше 130 °С; хімічно стійкими - зміна рН води після обробки ампул у стерилізаторі не повинна перевищувати 0,8.
Допускається виготовляти ампули з інших марок медичного скла, що не погіршують якість продукції. До першого класу відносять марки скла НС-3, НС-1, до другого - НС-2, АБ-1.
Виробництво ампул здійснюється на склозаводах зі скляних трубок (склодроту) вищенаведених класів і марок скла.
Всі типи ампул виготовляють із склодроту на роторних скло- формувальних автоматах або напівавтоматах різних фірм 10-8 «ТУНГСРАМ» (Угорщина), «АМБЕГ», «МАТВЕР» (Німеччина). Недоліком такого способу виготовлення ампул є утворення внутрішньої напруги, коли відбувається перерозподіл довжини зв’язків між молекулами у складі скла, що може призвести до механічного руйнування виробу або появи мікротріщин при несприятливих чинниках (висока температура, різка зміна температур, вібрація і т.д.), тому після виготовлення здійснюють зняття залишкової напруги за допомогою відпалювання ампул у спеціальних печах. Процес відпалювання полягає в нагріванні ампул або флаконів до температури, близької до температури розм’якшення скла, витримці їх при цій температурі протягом 7-10 хвилин і поступовому охолоджуванні.
Американською фірмою «Корнінг-глас» розроблено новий метод виготовлення ампул без проміжного використання дроту. Процес формування скловиробів на цих машинах відбувається струминно-видувним методом, який забезпечує високий ступінь рівномірності розподілу скломаси у стінках готових виробів.
До скла висуваються наступні вимоги:
^ прозорість - для візуального й оптичного контролю на відсутність механічних включень;
^ безбарвність - дозволяє виявляти, окрім механічних включень, зміну кольору розчину;
^ легкоплавкість - необхідна для якісного запаювання ампул при порівняно невисокій температурі, щоб уникнути нагрівання розчину;
^ термічна стійкість - здатність скляних виробів не руйнуватися при різких коливаннях температури;
^ хімічна стійкість, що гарантує збереження лікарських речовин та інших компонентів препарату, показує здатність скла до вилужу вання;
^ механічна міцність - для витримування навантажень при обробці ампул у процесі виробництва, транспортування і зберігання. Ця вимога повинна поєднуватися з необхідною крихкістю для легкого розкриття капіляра ампул.
Підготовка ампул до наповнення включає наступні операції: розкриття капілярів, визначення якості ампул, миття, сушка і (або) стерилізація ампул.
Якість ампульного скла і ампул оцінюють за наступними параметрами:
- Водостійкість.
- Лугостійкість.
- Залишкова напруга.
- Термічна стійкість.
- Хімічна стійкість.
- Світлозахисні властивості (для скла СНС-1).
- Візуальний контроль ампул.
- Радіальне биття стебла ампул відносно корпуса.
- Відхилення від округлості ампул.
- Для ампул вакуумного наповнення проводять визначення глибини розрідження з метою точного наповнення ампул за допомогою вакууму.
- Легкоплавкість.
Після визначення якості ампули піддають зовнішньому і внутрішньому миттю. Зовнішнє миття частіше здійснюють методом душу- вання або поєднують із внутрішнім миттям. Для внутрішнього миття використовують такі методи: шприцевий, вакуумні (турбовакуумний, вихровий, пароконденсаційний), вібраційний, термічний, ультразвукові (віброультразвуковий, контактно-ультразвуковиіі).
Як упаковка для парентеральних ліків усе ширше застосовуються полімери медичного призначення (поліетилен, полівінілхлоридний пластикат, фторопласт, поліпропілен і т.д.). їх переваги полягають насамперед у легкості, міцності, світлопроникності, широкій можливості використання для упаковок одноразового призначення.
Серед великого числа вживаних полімерів, проте, лише деякі витримують термічну стерилізацію без зміни своїх фізико-механічних властивостей. Ця обставина значною мірою стримує розповсюдження полімерів як матеріалів для упаковування ін’єкційних розчинів. Основні вимоги до упаковок з полімерів:
- безпека для здоров’я;
- достатня механічна міцність;
- можливість стерилізації в автоклаві;
- сумісність з лікарськими і допоміжними речовинами;
- зручність конструкції упаковок;
- непроникність для мікрофлори;
- постійність властивостей при температурних коливаннях у процесі зберігання;
- забезпечення стабільності і задовільна якість препаратів під час зберігання протягом двох років.